The Xylella fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS)

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
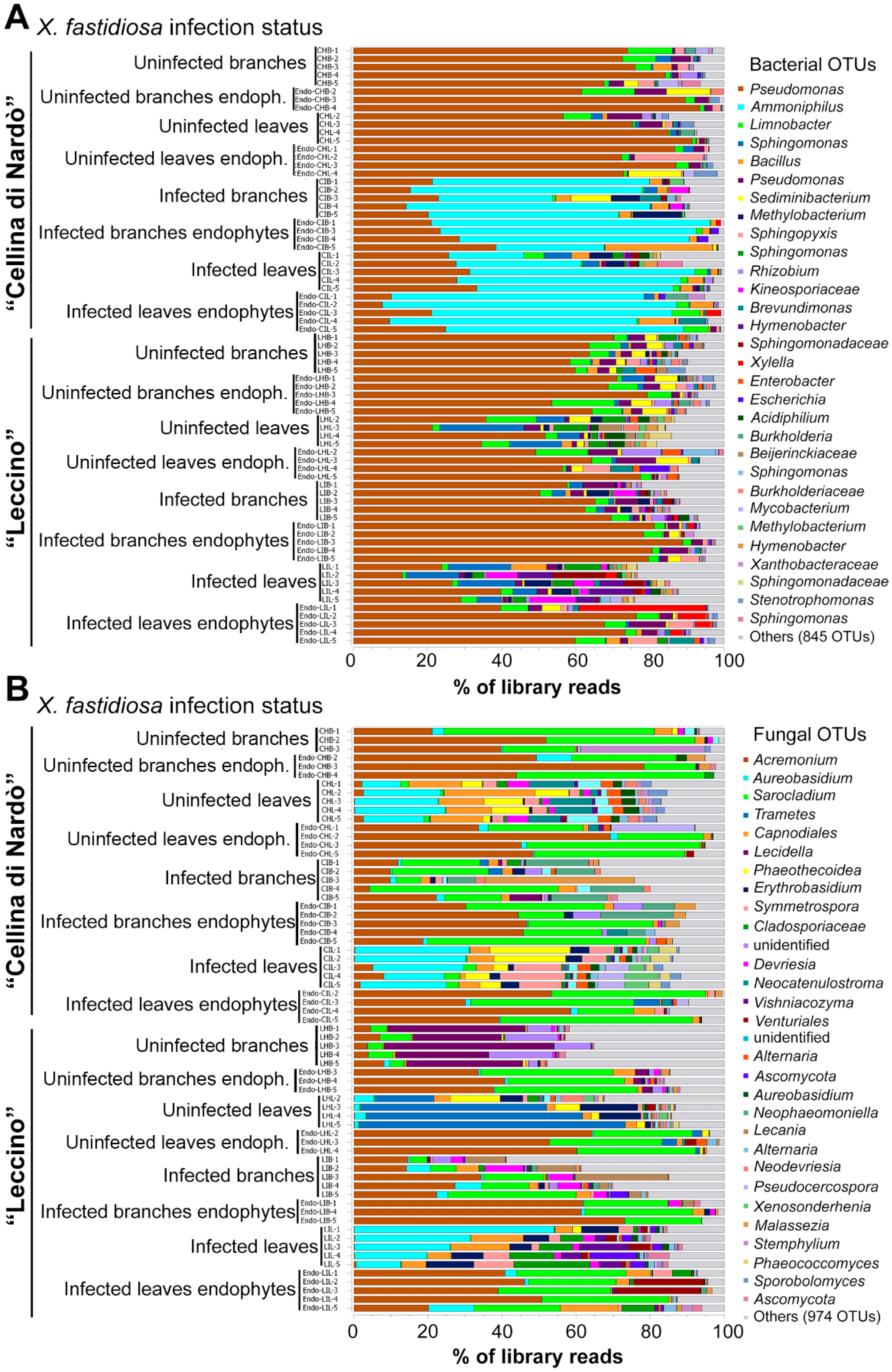
2.1. 16S rRNA Gene and ITS2 Illumina Sequencing Output
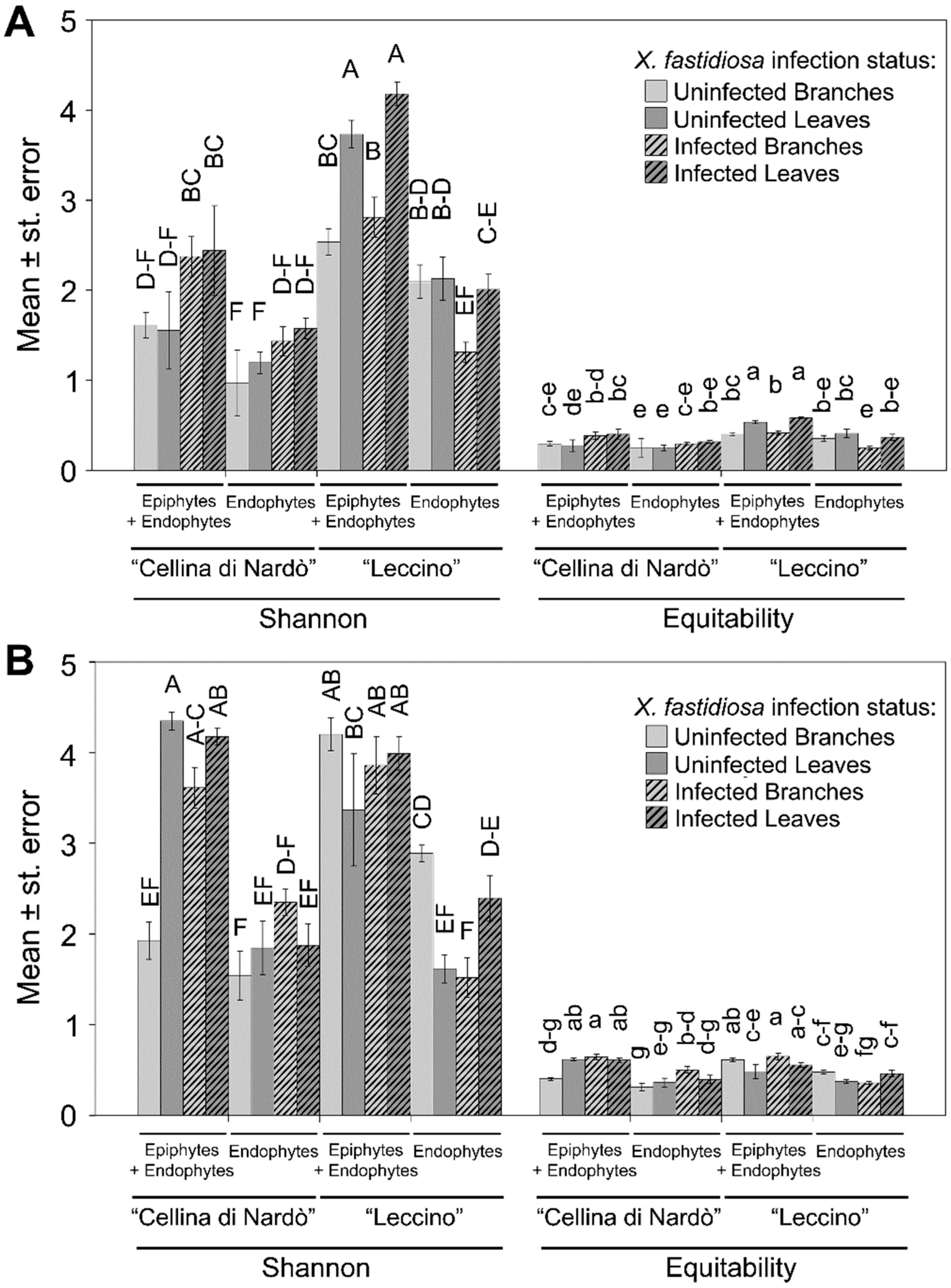
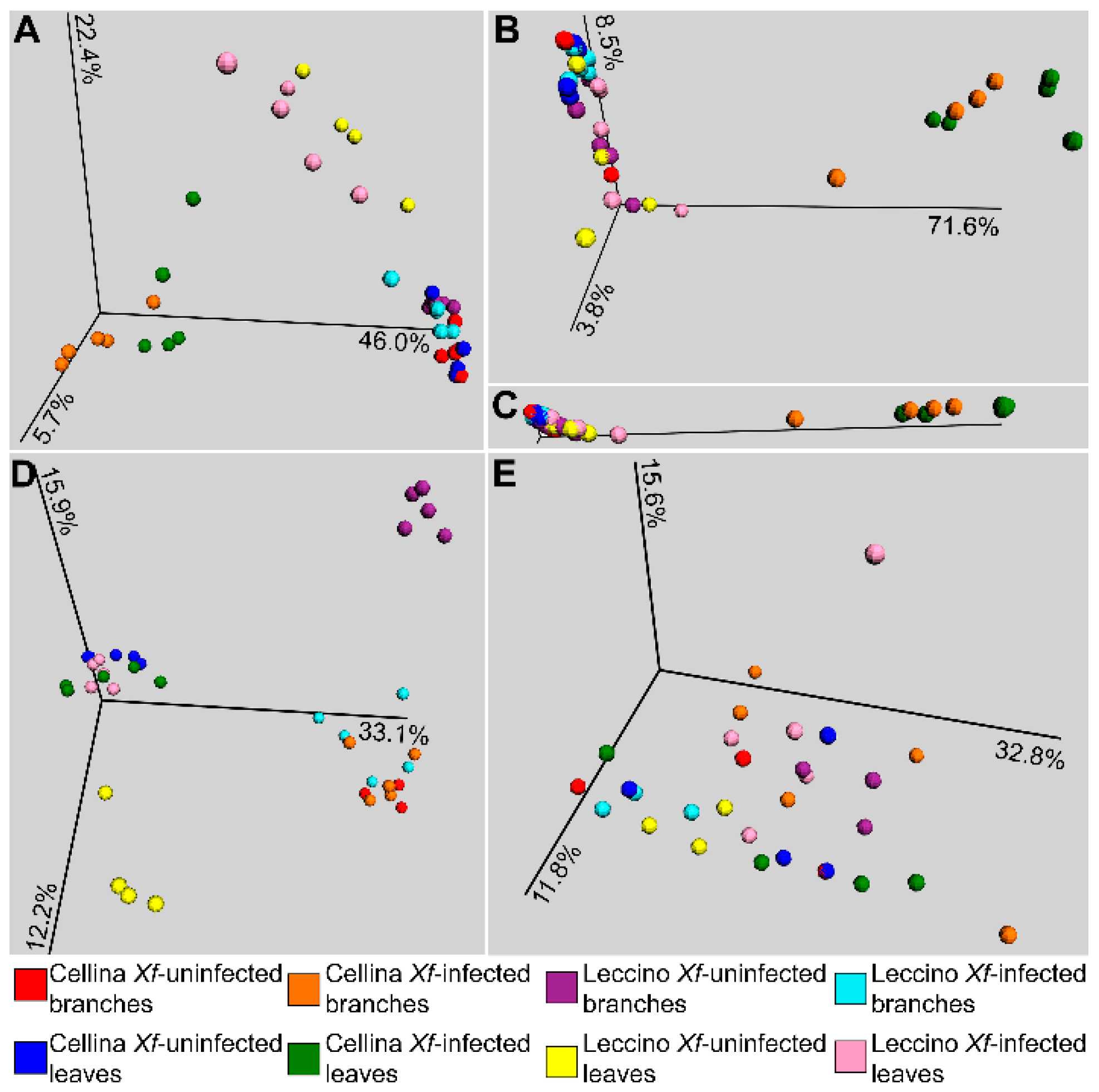
2.2. Sequence Analysis
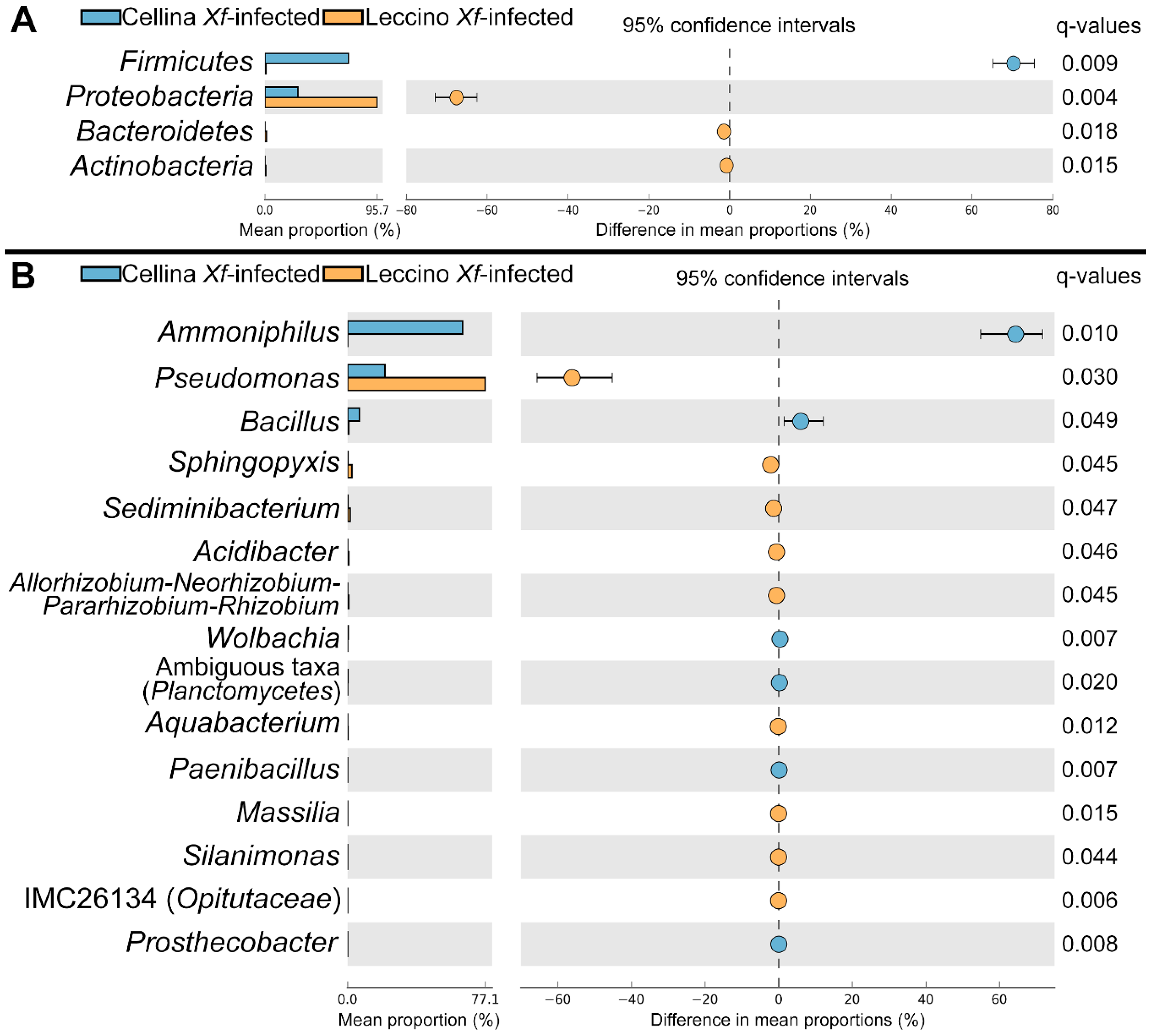
2.3. Identification of Significant Taxa
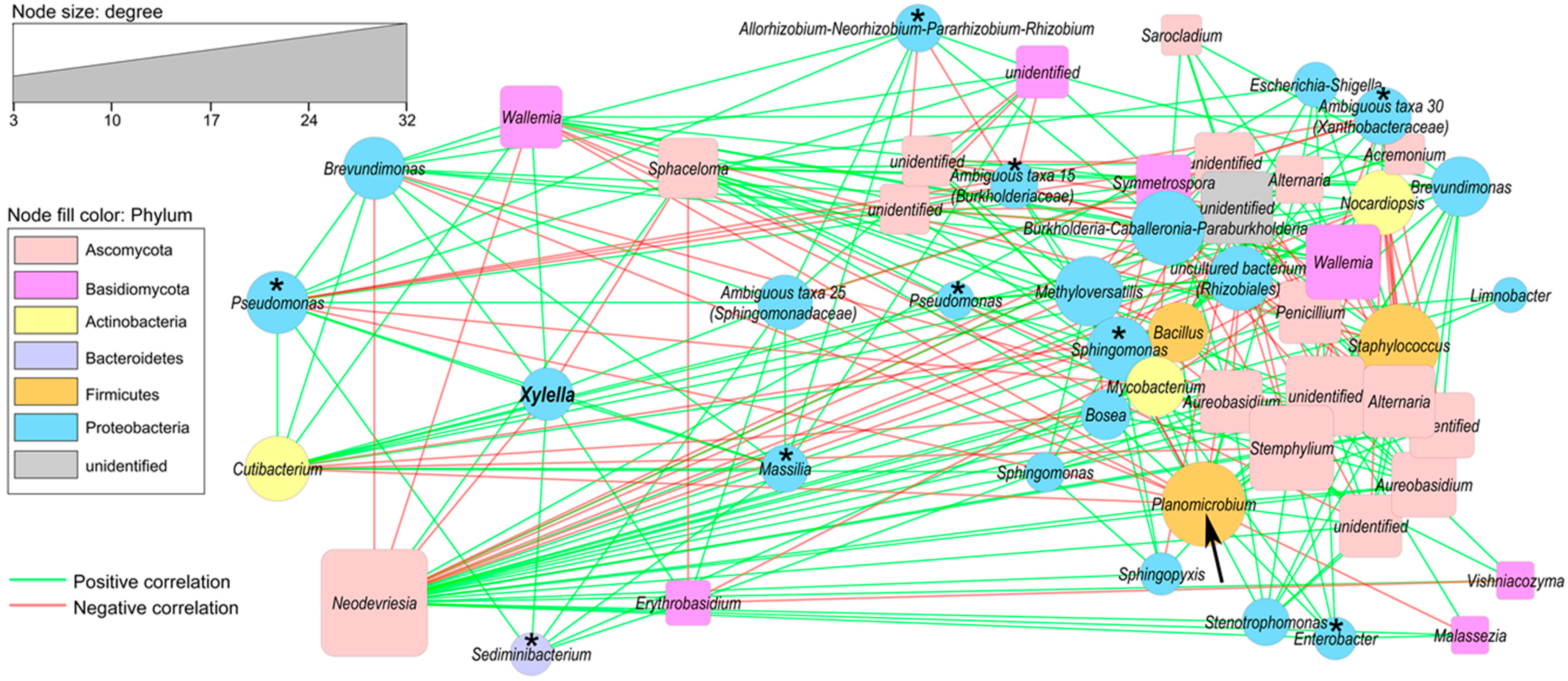
2.4. Correlation of Co–Occurrence Patterns and Network Analysis
3. Discussion
4. Methods
4.1. Sample Collection
4.2. DNA Extraction, PCR, and Illumina Sequencing of 16S rRNA Gene and ITS2 Amplicon Libraries
4.3. Bioinformatic Analysis of the Sequences
4.4. Correlation of Co–Occurrence Patterns and Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants. The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Gopal, M.; Gupta, A. Microbiome selection could spur next-generation plant breeding strategies. Front. Microbiol. 2016, 7, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, M.E.; Hampton-Marcell, J.; Zarraonaindia, I.; Owens, S.M.; Moreau, C.S.; Gilbert, J.A.; Hartsel, J.; Kennedy, S.J.; Gibbons, S.M. Understanding cultivar-specificity and soil determinants of the cannabis microbiome. PLoS ONE 2014, 9, e99641. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Loren Themaat, E.V.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kloepper, J.W.; Ryu, C.M. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009, 14, 1–4. [Google Scholar] [CrossRef]
- Partida-Martínez, L.P.; Heil, M. The microbe-free plant: Fact or artifact? Front. Plant Sci. 2011, 2, 100. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.; Berg, C.; Landa, B.B.; Auerbach, A.; Moissl-Eichinger, C.; Berg, G. Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees. Front. Microbiol. 2015, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding analysis of fungal diversity in the phyllosphere and carposphere of olive (Olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef] [Green Version]
- Preto, G.; Martins, F.; Pereira, J.A.; Baptista, P. Fungal community in olive fruits of cultivars with different susceptibilities to anthracnose and selection of isolates to be used as biocontrol agents. Biol. Control 2017, 110, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Soussi, A.; Ferjani, R.; Marasco, R.; Guesmi, A.; Cherif, H.; Rolli, E.; Mapelli, F.J.; Ouzari, H.I.; Daffonchio, D.; Cherif, A. Plant-associated microbiomes in arid lands. Diversity, ecology and biotechnological potential. Plant Soil 2016, 405, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Lama Cabanás, C.; Legarda, G.; Ruano-Rosa, D.; Pizarro-Tobías, P.; Valverde-Corredor, A.; Niqui, J.L.; Triviño, J.C.; Roca, A.; Mercado-Blanco, J. Indigenous Pseudomonas spp. Strains from the Olive (Olea europaea L.) rhizosphere as effective biocontrol agents against Verticillium dahliae: From the host roots to the bacterial genomes. Front. Microbiol. 2018, 9, 277. [Google Scholar] [CrossRef] [Green Version]
- Passos da Silva, D.; Castañeda-Ojeda, M.P.; Moretti, C.; Buonaurio, R.; Ramos, C.; Venturi, V. Bacterial multispecies studies and microbiome analysis of a plant disease. Microbiology 2014, 160, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Saponari, M.; Boscia, D.; Altamura, G.; Loconsole, G.; Zicca, S.; D’Attoma, G.; Morelli, M.; Palmisano, F.; Saponari, A.; Tavano, D.; et al. Isolation and pathogenicity of Xylella fastidiosa associated to the olive quick decline syndrome in southern Italy. Sci. Rep. 2017, 7, 17723. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, G.; Semeraro, T.; Aretano, R.; De Bellis, L.; Luvisi, A. GIS analysis of land-use change in threatened landscapes by Xylella fastidiosa. Sustainability 2019, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Saponari, M.; Loconsole, G.; Cornara, D.; Yokomi, R.K.; De Stradis, A.; Boscia, D.; Bosco, D.; Martelli, G.P.; Krugner, R.; Porcelli, F. Infectivity and transmission of Xylella fastidiosa by Philaenus spumarius (Hemiptera: Aphrophoridae) in Apulia, Italy. J. Econ. Entomol. 2014, 107, 1316–1319. [Google Scholar] [CrossRef] [Green Version]
- Strona, G.; Carstens, C.J.; Beck, P.S.A. Network analysis reveals why Xylella fastidiosa will persist in Europe. Sci. Rep. 2017, 7, 71. [Google Scholar] [CrossRef]
- EFSA. Update of a database of host plants of Xylella fastidiosa: 20 November 2015. EFS2 J. 2016, 14. [Google Scholar] [CrossRef]
- Carlucci, A.; Lops, F.; Marchi, G.; Mugnai, L.; Surico, G. Has Xylella fastidiosa "chosen" olive trees to establish in the Mediterranean basin? Phytopathol. Mediterr. 2013, 52, 541–544. [Google Scholar]
- Luvisi, A.; Ampatzidis, Y.; De Bellis, L. Plant pathology and information technology: Opportunity for management of disease outbreak and applications in regulation frameworks. Sustainability 2016, 8, 831. [Google Scholar] [CrossRef] [Green Version]
- Bucci, E.M. Xylella fastidiosa, a new plant pathogen that threatens global farming: Ecology, molecular biology, search for remedies. Biochem. Biophys. Res. Commun. 2018, 502, 173–182. [Google Scholar] [CrossRef] [PubMed]
- McElrone, A.J.; Jackson, S.; Habdas, P. Hydraulic disruption and passive migration by a bacterial pathogen in oak tree xylem. J. Exp. Bot. 2018, 59, 2649–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Sun, Y.; Walker, M.A.; Labavitch, J.M. Vascular occlusions in grapevines with Pierce’s disease make disease symptom development worse. Plant Physiol. 2013, 161, 1529–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, M.; Luvisi, A.; Meyer, J.B.; Sabella, E.; De Bellis, L.; Cruz, A.C.; Ampatzidis, Y.; Cherubini, P. Specific Fluorescence in Situ Hybridization (FISH) test to highlight colonization of Xylem vessels by Xylella fastidiosa in naturally infected olive trees (Olea europaea L.). Front. Plant Sci. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Benedictis, M.; De Caroli, M.; Baccelli, I.; Marchi, G.; Bleve, G.; Gallo, A.; Ranaldi, F.; Falco, V.; Pasquali, V.; Piro, G.; et al. Vessel occlusion in three cultivars of Olea europaea naturally exposed to Xylella fastidiosa in open field. J. Phytopathol. 2017, 165, 589–594. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Morelli, M.; Saponari, M.; Loconsole, G.; Chiumenti, M.; Boscia, D.; Savino, V.N.; Martelli, G.P.; Saldarelli, P. Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp. pauca. BMC Genom. 2016, 1, 475. [Google Scholar] [CrossRef] [Green Version]
- Martelli, G.P. The current status of the quick decline syndrome of olive in southern Italy. Phytoparasitica 2016, 44, 1–10. [Google Scholar] [CrossRef]
- Luvisi, A.; Aprile, A.; Sabella, E.; Vergine, M.; Nicolì, F.; Nutricati, E.; Miceli, A.; Negro, C.; De Bellis, L. Xylella fastidiosa subsp. pauca (CoDiRO strain) infection in four olive (Olea europaea L.) cultivars: Profile of phenolic compounds in leaves and progression of leaf scorch symptoms. Phytopathol. Mediterr. 2017, 56, 259–273. [Google Scholar]
- Sabella, E.; Luvisi, A.; Aprile, A.; Negro, C.; Vergine, M.; Nicolì, F.; Miceli, A.; De Bellis, L. Xylella fastidiosa induces differential expression of lignification related-genes and lignin accumulation in tolerant olive trees cv. Leccino. J. Plant Physiol. 2018, 220, 60–68. [Google Scholar] [CrossRef]
- Deyett, E.; Roper, M.C.; Ruegger, P.; Yang, J.I.; Borneman, J.; Rolshausen, P.E. Microbial landscape of the grapevine endosphere in the context of pierce’s disease. Phytobiomes 2017, 1, 138–149. [Google Scholar] [CrossRef]
- Azevedo, J.L.; Araújo, W.L.; Lacava, P.T. The diversity of citrus endophytic bacteria and their interactions with Xylella fastidiosa and host plants. Gen. Mol. Biol. 2016, 39, 476–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, P.; Cameirão, C.; Giampetruzzi, A.; Morelli, M.; Kubaa, R.A.; Altamura, G.; D’Attoma, G.; Pereira, J.A.; Neto, T.L.; Saldarelli, P. Understanding the olive microbiome of susceptible and resistant cultivars for sustainable biocontrol. J. Plant Pathol. 2019, 101, 849. [Google Scholar] [CrossRef]
- Berg, G.; Köberl, M.; Rybakova, D.; Müller, H.; Grosch, R.; Smalla, K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Sheflin, A.M.; Manter, D.K.; Vivanco, J.M. Manipulating the soil microbiome to increase soil health and plant fertility. Biol. Fertil. Soils 2012, 48, 489–499. [Google Scholar] [CrossRef]
- Ramírez-Puebla, S.T. Servín-Garcidueñas, L.E.; Jiménez-Marín, B.; Bolaños, L.M.; Rosenblueth, M.; Martínez, J.; Rogel, M.A.; Ormeño-Orrillo, E.; Martínez-Romero, E. Gut and root microbiota commonalities. Appl. Environ. Microbiol. 2013, 79, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Saponari, M.; Boscia, D.; Nigro, F.; Martelli, G.P. Identification of DNA sequences related to Xylella fastidiosa in oleander, almond and olive trees exhibiting leaf scorch symptoms in Apulia (southern Italy). J. Plant Pathol. 2013, 95, 668. [Google Scholar]
- Bragard, C.; Dehnen-Schmutz, K.; Di Serio, F.; Gonthier, P.; Jacques, M.A.; Miret, J.A.J.; Fejer Justesen, A.; MacLeod, A.; Magnusson, C.S.; Milonas, P.; et al. Effectiveness of in planta control measures for Xylella fastidiosa. EFS2 J. 2019, 17, 459. [Google Scholar] [CrossRef]
- Morelli, M.; D’Attoma, G.; Saponari, M.; Saldarelli, P. Evaluation of an endophytic symbiont as a putative biocontrol agent of the codiro strain of Xylella fastidiosa. J. Plant Pathol. 2019, 99, S54. [Google Scholar] [CrossRef]
- Cruz, A.C.; Luvisi, A.; De Bellis, L.; Ampatzidis, Y. X-FIDO: An effective application for detecting olive quick decline syndrome with deep learning and data fusion. Front. Plant Sci. 2017, 8, 1741. [Google Scholar] [CrossRef]
- Chiriacò, M.S.; Luvisi, A.; Primiceri, E.; Sabella, E.; De Bellis, L.; Maruccio, G. Development of a lab-on-a-chip method for rapid assay of Xylella fastidiosa subsp. pauca strain CoDiRO. Sci. Rep. 2018, 8, 7376. [Google Scholar] [CrossRef]
- Redford, A.J.; Bowers, R.M.; Knight, R.; Linhart, Y.; Fierer, N. The ecology of the phyllosphere: Geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 2010, 12, 2885–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, B.; Rime, T.; Phillips, M.; Stierli, B.; Hajdas, I.; Widmer, F.; Hartmann, M. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 2016, 92, fiw018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitsev, G.M.; Tsitko, I.V.; Rainey, F.A.; Trotsenko, Y.A.; Uotila, J.S.; Stackebrandt, E.; Salkinoja-Salonen, M.S. New aerobic ammonium-dependent obligately oxalotrophic bacteria: Description of Ammoniphilus oxalaticus gen. nov., sp. nov. and Ammoniphilus oxalivorans gen. nov., sp. nov. Int. J. Syst. Bacteriol. 1998, 48 Pt 1, 151–163. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Gough, E.; Shaikh, H.; Manges, A.R. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin. Infect. Dis. 2011, 53, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Sánchez-Cañizares, C.; Jorrín, B.; Poole, P.S.; Tkacz, A. Understanding the holobiont: The interdependence of plants and their microbiome. Curr. Opin. Microbiol. 2017, 38, 188–196. [Google Scholar] [CrossRef]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100. [Google Scholar] [CrossRef]
- Doty, S.L. (Ed.) Functional Importance of the Plant Microbiome; Springer: Cham, Switzerland, 2017; pp. 1–5. [Google Scholar]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Jeffries, T.C.; Gaitan, J.J.; Encinar, D.; Berdugo, M.; Campbell, C.D.; Singh, B.K. Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 2016, 7, 10541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Elsas, J.D.; Chiurazzi, M.; Mallon, C.A.; Elhottovā, D.; Krištůfek, V.; Falcão Salles, J. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. USA 2012, 109, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehner, J.; Antunes, P.M.; Powell, J.R.; Mazukatow, J.; Rillig, M.C. Plant pathogen protection by arbuscular mycorrhizas: A role for fungal diversity? Pedobiologia 2010, 53, 197–201. [Google Scholar] [CrossRef]
- van der Heijden, M.G.A.; Wagg, C. Soil microbial diversity and agro-ecosystem functioning. Plant Soil 2013, 363, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Denison, R.F.; Toby Kiers, E. Why are most rhizobia beneficial to their plant hosts, rather than parasitic? Microbes Infect. 2004, 6, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Spann, T.; Wang, N. Isolation and characterization of beneficial bacteria associated with citrus roots in Florida. Microb. Ecol. 2011, 62, 324–336. [Google Scholar] [CrossRef]
- Khan, A.L.; Waqas, M.; Kang, S.M.; Al-Harrasi, A.; Hussain, J.; Al-Rawahi, A.; Al-Khiziri, S.; Ullah, I.; Ali, L.; Jung, H.Y.; et al. Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J. Microbiol. 2014, 52, 689–695. [Google Scholar] [CrossRef]
- Naveed, M.; Joubert, P.M.; Doty, S.L. The endophyte Enterobacter sp. FD17: A maize growth enhancer selected based on rigorous testing of plant beneficial traits and colonization characteristics. Biol. Fertil. Soil 2014, 50, 249–262. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Cardinale, M.; Suarez, C.; Steffens, D.; Ratering, S.; Schnell, S. Effect of different soil phosphate sources on the active bacterial microbiota is greater in the rhizosphere than in the endorhiza of barley (Hordeum vulgare L.). Microb. Ecol. 2018, 77, 689–700. [Google Scholar] [CrossRef]
- Mercado-Blanco, J.; Bakker, P.A.H.M. Interactions between plants and beneficial Pseudomonas spp.: Exploiting bacterial traits for crop protection. Antonie Van Leeuwenhoek 2007, 92, 367–389. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Bulgarelli, D. The plant microbiome at work. Mol. Plant Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deines, P.; Bosch, T.C.G. Transitioning from microbiome composition to microbial community interactions: The potential of the metaorganism hydra as an experimental model. Front. Microbiol. 2016, 7, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [Green Version]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of ’unculturable’ bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, M.; Grube, M.; Erlacher, A.; Quehenberger, J.; Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 2015, 17, 239–252. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere. High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Ambika Manirajan, B.; Maisinger, C.; Ratering, S.; Rusch, V.; Schwiertz, A.; Cardinale, M.; Schnell, S. Diversity, specificity, co-occurrence and hub taxa of the bacterial–fungal pollen microbiome. FEMS Microbiol. Ecol. 2018, 94, fiy112. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Nicolì, F.; Negro, C.; Nutricati, E.; Vergine, M.; Aprile, A.; Sabella, E.; Damiano, G.; De Bellis, L.; Luvisi, A. Accumulation of azelaic acid in Xylella fastidiosa-infected olive trees: A mobile metabolite for health screening. Phytopathology 2018, 109, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ministero dei beni e delle attività culturali e del turismo; Direzione regionale per i beni culturali e paesaggistici della Puglia; Regione Puglia. Integrazione alle dichiarazioni di notevole interesse pubblico della fascia costiera salentina, ricadente nei comuni di Lecce, Vernole, Melendugno e Otranto. Available online: http://paesaggio.regione.puglia.it/PPTR_2015/6_%20Il%20sistema%20delle%20tutele/6.4_Schede%20di%20identificazione%20e%20discipline%20d’uso/PAE0134_scheda.pdf (accessed on 16 December 2019).
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Romanazzi, G.; Murolo, S.; Pizzichini, L.; Nardi, S. Esca in young and mature vineyards, and molecular diagnosis of the associated fungi. Eur. J. Plant Pathol. 2009, 125, 277–290. [Google Scholar] [CrossRef]
- Garrido, C.; Carbú, M.F.; Fernández-Acero, J.; Boonham, N.; Colyer, A.J.; Cantoral, M.; Budge, G. Development of protocols for detection of Colletotrichum acutatum and monitoring of strawberry anthracnose using real-time PCR. Plant Pathol. 2009, 58, 43–51. [Google Scholar] [CrossRef]
- Martín, M.T.; Cuesta, M.J.; Martín, L. Development of SCAR primers for PCR assay to detect Diplodia seriata. Int. Sch. Res. Not. 2014, 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.T.; Cobos, R.; Martín, L.; López-Enríquez, L. Real-time PCR detection of Phaeomoniella chlamydospora and Phaeoacremonium aleophilum. Appl. Environ. Microbiol. 2012, 78, 3985–3991. [Google Scholar] [CrossRef] [Green Version]
- Aroca, A.; Raposo, R.; Lunello, P. A biomarker for the identification of four Phaeoacremonium species using the beta-tubulin gene as the target sequence. Appl. Microbiol. Biotechnol. 2008, 80, 1131–1140. [Google Scholar] [CrossRef]
- Carlucci, A.; Lops, F.; Cibelli, F.; Raimondo, M.L. Phaeoacremonium species associated with olive wilt and decline in southern Italy. Eur. J. Plant Pathol. 2015, 141, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Drenth, A.; Wagels, G.; Smith, B.; Sendall, B.; O’Dwyer, C.; Irvine, G.; Irwin, J.A.G. Development of a DNA-based method for detection and identification of Phytophthora species. Australas. Plant Path. 2006, 35, 147–159. [Google Scholar] [CrossRef]
- Bilodeau, G.J.; Koike, S.T.; Uribe, P.; Martin, F.N. Development of an assay for rapid detection and quantification of Verticillium dahliae in soil. Phytopathology 2012, 102, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]
- D’Attoma, G.; Morelli, M.; Saldarelli, P.; Saponari, M.; Giampetruzzi, A.; Boscia, D.; Savino, V.N.; De La Fuente, L.; Cobine, P.A. Ionomic differences between susceptible and resistant olive cultivars infected by Xylella fastidiosa in the outbreak area of salento, italy. Pathogens 2019, 8, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for illumina amplicon sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef]
- Mayerhofer, J.; Eckard, S.; Hartmann, M.; Grabenweger, G.; Widmer, F.; Leuchtmann, A.; Enkerli, J. Assessing effects of the entomopathogenic fungus Metarhizium brunneum on soil microbial communities in Agriotes spp. biological pest control. FEMS Microbiol. Ecol. 2017, 93, fix117. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sanchez-Garcia, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH. A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Jackson, M.A.; Bell, J.T.; Spector, T.D.; Steves, C.J. A heritability-based comparison of methods used to cluster 16S rRNA gene sequences into operational taxonomic units. PeerJ 2016, 4, e2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project. Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Henrik Nilsson, R.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi-recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data. A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror. A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.; Beiko, R. Encyclopedia of Metagenomics; Nelson, K.E., Ed.; Springer Science + Business Media: New York, NY, USA, 2013; pp. 1–6. [Google Scholar]
- White, H. A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica 1980, 48, 817–838. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Robertson, C.E.; Harris, J.K.; Wagner, B.D.; Granger, D.; Browne, K.; Tatem, B.; Feazel, L.M.; Park, K.; Pace, N.R.; Frank, D.N. Explicet: Graphical user interface software for metadata-driven management, analysis and visualization of microbiome data. Bioinformatics 2013, 29, 3100–3101. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape. A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Kawai, S. An algorithm for drawing general undirected graphs. Inf. Process. Lett. 1989, 31, 7–15. [Google Scholar] [CrossRef]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape. Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Agler, M.T.; Ruhe, J.; Constanze Morhenn, S.K.; Kim, S.T.; Weigel, D.; Keme, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p Values | |||||
|---|---|---|---|---|---|
| Bacteria | Fungi | ||||
| Factor | Subject | Epiphytes + Endophytes | Endo-phytes | Epiphytes + Endophytes | Endo-Phytes |
| Infection status | All samples | 0.001 | 0.001 | 0.024 | 0.786 |
| Infection status | “Cellina di Nardò” | 0.001 | 0.001 | 0.069 | 0.446 |
| Infection status | “Leccino” | 0.067 | 0.069 | 0.001 | 0.184 |
| Infection status | “Cellina di Nardò” Branches | 0.001 | 0.028 | 0.001 | 0.369 |
| Infection status | “Leccino” Branches | 0.007 | 0.012 | 0.001 | 0.0014 |
| Infection status | “Cellina di Nardò” Leaves | 0.001 | 0.001 | 0.001 | 0.902 |
| Infection status | “Leccino” Leaves | 0.035 | 0.429 | 0.006 | 0.114 |
| Cultivar | All samples | 0.002 | 0.001 | 0.004 | 0.081 |
| Cultivar | Xf-infected | 0.001 | 0.001 | 0.028 | 0.05 |
| Cultivar | Xf-uninfected | 0.001 | 0.040 | 0.001 | 0.276 |
| Cultivar | Xf-infected Branches | 0.001 | 0.009 | 0.001 | 0.055 |
| Cultivar | Xf-infected Leaves | 0.001 | 0.013 | 0.001 | 0.122 |
| Cultivar | Xf-uninfected Branches | 0.011 | 0.243 | 0.001 | 0.0014 |
| Cultivar | Xf-uninfected Leaves | 0.001 | 0.101 | 0.001 | 0.32 |
| Habitat | All samples | 0.014 | 0.483 | 0.001 | 0.205 |
| Habitat | “Cellina di Nardò” | 0.874 | 0.723 | 0.001 | 0.264 |
| Habitat | “Leccino” | 0.001 | 0.061 | 0.001 | 0.256 |
| Habitat | Xf-infected | 0.138 | 0.368 | 0.001 | 0.207 |
| Habitat | Xf-uninfected | 0.020 | 0.763 | 0.001 | 0.271 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergine, M.; Meyer, J.B.; Cardinale, M.; Sabella, E.; Hartmann, M.; Cherubini, P.; De Bellis, L.; Luvisi, A. The Xylella fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS). Pathogens 2020, 9, 35. https://doi.org/10.3390/pathogens9010035
Vergine M, Meyer JB, Cardinale M, Sabella E, Hartmann M, Cherubini P, De Bellis L, Luvisi A. The Xylella fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS). Pathogens. 2020; 9(1):35. https://doi.org/10.3390/pathogens9010035
Chicago/Turabian StyleVergine, Marzia, Joana B. Meyer, Massimiliano Cardinale, Erika Sabella, Martin Hartmann, Paolo Cherubini, Luigi De Bellis, and Andrea Luvisi. 2020. "The Xylella fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS)" Pathogens 9, no. 1: 35. https://doi.org/10.3390/pathogens9010035